User guide¶
Parameters¶
To view command-line parameters type miRge3.0 -h:
usage: miRge3.0 [options]
miRge3.0 (Comprehensive analysis of small RNA sequencing Data)
optional arguments:
-h, --help show this help message and exit
--version show program's version number and exit
Options:
-s, --samples list of one or more samples separated by comma or a file with list of samples separated by new line (accepts *.fastq, *.fastq.gz)
-db, --mir-DB the reference database of miRNA. Options: miRBase and miRGeneDB (Default: miRBase)
-lib, --libraries-path the path to miRge libraries
-on, --organism-name the organism name can be human, mouse, fruitfly, nematode, rat or zebrafish
-ex, --crThreshold the threshold of the proportion of canonical reads for the miRNAs to retain. Range for ex (0 - 0.5), (Default: 0.1)
-phr, --phred64 phred64 format (Default: 33)
-spk, --spikeIn switch to annotate spike-ins if spike-in bowtie index files are located at the path of bowtie's index files (Default: off)
-ie, --isoform-entropy switch to calculate isomir entropy (default: off)
-cpu, --threads the number of processors to use for trimming, qc, and alignment (Default: 1)
-ai, --AtoI switch to calculate A to I editing (Default: off)
-tcf --tcf-out switch to write trimmed and collapsed fasta file (Default: off)
-gff --gff-out switch to output isomiR results in gff format (Default: off)
-bam --bam-out switch to output isomiR results in gff format (Default: off)
-trf --tRNA-frag switch to analyze tRNA fragment and halves (Default: off)
-o --outDir the directory of the outputs (Default: current directory)
-shh --quiet enable quiet/silent mode, only show warnings and errors (Default: off)
Data pre-processing:
-a, --adapter Sequence of a 3' adapter. The adapter and subsequent bases are trimmed
-g, --front Sequence of a 5' adapter. The adapter and any preceding bases are trimmed
-u, --cut Remove bases from each read. If LENGTH is positive, remove bases from the beginning. If LENGTH is negative, remove bases from the end
-nxt, --nextseq-trim NextSeq-specific quality trimming (each read). Trims also dark cycles appearing as high-quality G bases
-q, --quality-cutoff Trim low-quality bases from 5' and/or 3' ends of each read before adapter removal. If one value is given, only the 3' end is trimmed
If two comma-separated cutoffs are given, the 5' end is trimmed with the first cutoff, the 3' end with the second
-l, --length Shorten reads to LENGTH. Positive values remove bases at the end while negative ones remove bases at the beginning. This and the following
modifications are applied after adapter trimming
-NX, --trim-n Trim N's on ends of reads
-m, --minimum-length Discard reads shorter than LEN. (Default: 16)
-umi, --uniq-mol-ids Removes PCR duplicates and trim UMI of length by specifying two comma-separated cutoffs as 5’ cutoff,3’ bp from both ends of the read. eg: 4,4 or 0,4
-udd, --umiDedup Specifies argument to removes PCR duplicates (Default: False); if TRUE it will remove UMI and remove PCR duplicates otherwise it only remove UMI and keep the raw counts
-umiq, --umiqiagen Removes PCR duplicates of reads obtained from Qiagen platform (Default: Illumina; "-umi x,y " Required)
Predicting novel miRNAs:
The predictive model for novel miRNA detection is trained on human and mouse!
-nmir, --novel-miRNA include prediction of novel miRNAs
-minl, --minLength the minimum length of the reatined reads for novel miRNA detection (default: 16)
-maxl, --maxLength the maximum length of the reatined reads for novel miRNA detection (default: 25)
-c, --minReadCounts the minimum read counts supporting novel miRNA detection (default: 2)
-mloc, --maxMappingLoci the maximum number of mapping loci for the retained reads for novel miRNA detection (default: 3)
-sl, --seedLength the seed length when invoking Bowtie for novel miRNA detection (default: 25)
-olc, --overlapLenCutoff the length of overlapped seqence when joining reads into longer sequences based on the coordinate
on the genome for novel miRNA detection (default: 14)
-clc, --clusterLength the maximum length of the clustered sequences for novel miRNA detection (default: 30)
Optional PATH arguments:
-pbwt, --bowtie-path the path to system's directory containing bowtie binary
-psam, --samtools-path the path to system's directory containing samtools binary
-prf, --RNAfold-path the path to system's directory containing RNAfold binary
miRge3.0 libraries¶
miRge3.0 pipeline aligns the raw reads against a set of small-RNA annotation libraries. The libraries specific to the organism of interest can be obtained from SourceForge. Downloading the libraries on terminal:
Command-line Interface (CLI)¶
We recommend to create a directory miRge3_Lib and download using wget as shown below,
mkdir miRge3_Lib
cd miRge3_Lib
wget -O human.tar.gz "https://sourceforge.net/projects/mirge3/files/miRge3_Lib/human.tar.gz/download"
wget -O mouse.tar.gz "https://sourceforge.net/projects/mirge3/files/miRge3_Lib/mouse.tar.gz/download"
wget -O rat.tar.gz "https://sourceforge.net/projects/mirge3/files/miRge3_Lib/rat.tar.gz/download"
wget -O nematode.tar.gz "https://sourceforge.net/projects/mirge3/files/miRge3_Lib/nematode.tar.gz/download"
wget -O fruitfly.tar.gz "https://sourceforge.net/projects/mirge3/files/miRge3_Lib/fruitfly.tar.gz/download"
wget -O zebrafish.tar.gz "https://sourceforge.net/projects/mirge3/files/miRge3_Lib/zebrafish.tar.gz/download"
wget -O hamster.tar.gz "https://sourceforge.net/projects/mirge3/files/miRge3_Lib/hamster.tar.gz/download"
Users can download only what is necessary. Unzip the files once downloaded by the following command:
tar -xzf human.tar.gz
Replace human with the organism of interest. If you want to extract all the files at once, you could use tar -xzf *.tar.gz instead.
Graphical User Interface (GUI)¶
We recommend to create a folder miRge3_Lib and download the libraries directly from SourceForge. Once downloaded, extract/unzip the compressed files.
Building new libraries¶
If you are interested in creating specific library for an organism that is not part of this set then please refer to miRge3_build.
CLI - Example usage¶
Example command usage:
miRge3.0 -s SRR772403.fastq,SRR772404.fastq,SRR772405.fastq,SRR772406.fastq -lib miRge3_Lib -on human -db mirgenedb -o output_dir -gff -nmir -trf -ai -cpu 12 -a illumina
Output command line:
bowtie version: 1.2.3
Samtools version: 1.7
RNAfold version: 2.4.14
Collecting and validating input files...
miRge3.0 will process 4 out of 4 input file(s).
Cutadapt finished for file SRR772403 in 2.5358 second(s)
Collapsing finished for file SRR772403 in 0.0126 second(s)
Cutadapt finished for file SRR772404 in 7.3542 second(s)
Collapsing finished for file SRR772404 in 0.2786 second(s)
Cutadapt finished for file SRR772405 in 11.0667 second(s)
Collapsing finished for file SRR772405 in 0.8585 second(s)
Cutadapt finished for file SRR772406 in 3.5771 second(s)
Collapsing finished for file SRR772406 in 0.8677 second(s)
Matrix creation finished in 0.3838 second(s)
Data pre-processing completed in 27.2443 second(s)
Alignment in progress ...
Alignment completed in 15.8305 second(s)
Summarizing and tabulating results...
The number of A-to-I editing sites for is less than 10 so that no heatmap is drawn.
Summary completed in 71.4691 second(s)
Predicting novel miRNAs
Performing prediction of novel miRNAs...
Start to predict
Prediction of novel miRNAs Completed (104.83 sec)
The analysis completed in 222.2487 second(s)
miRge3.0 GUI¶
The application is cross platform, the image below is a screenshot of the software from MacOS
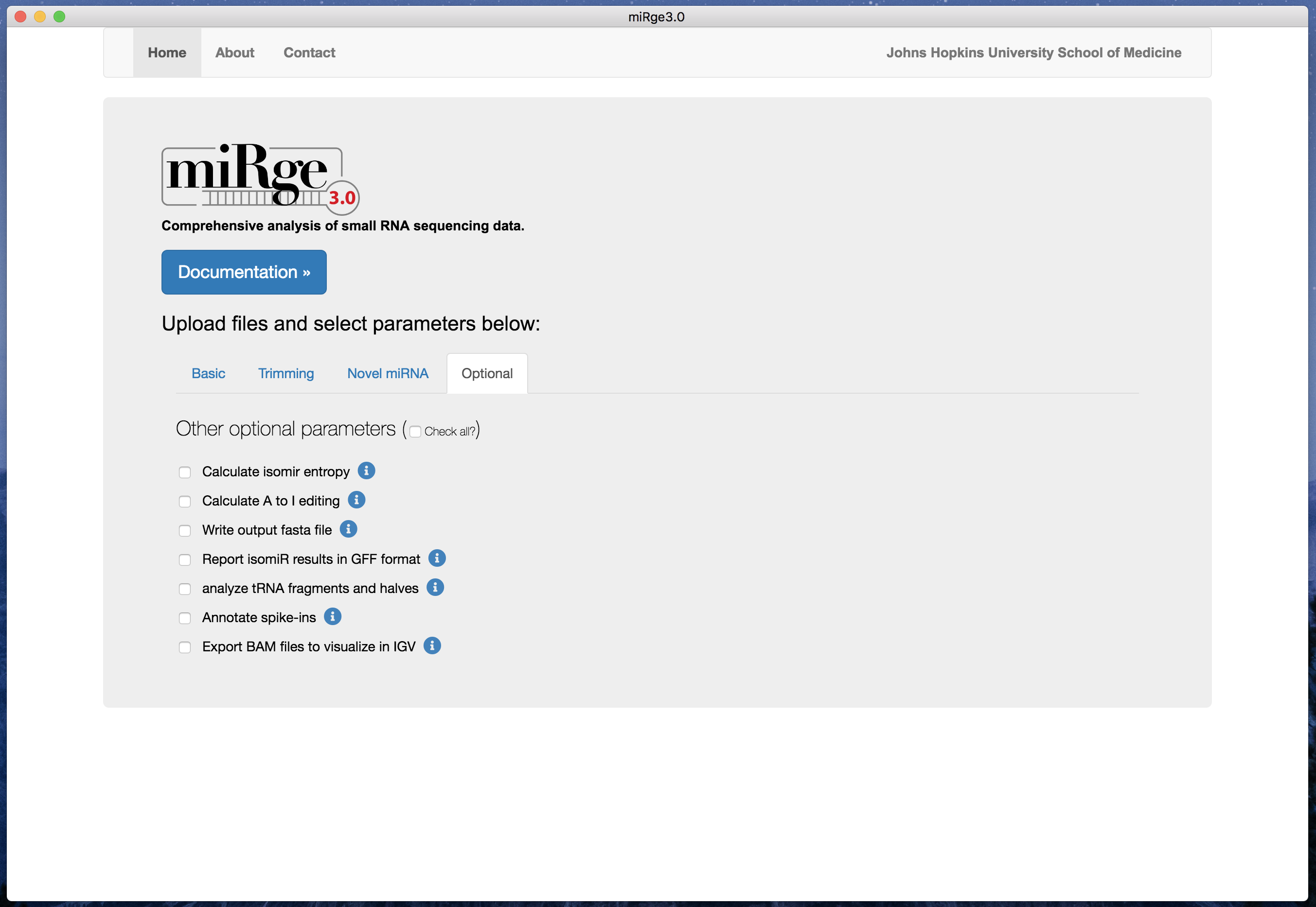
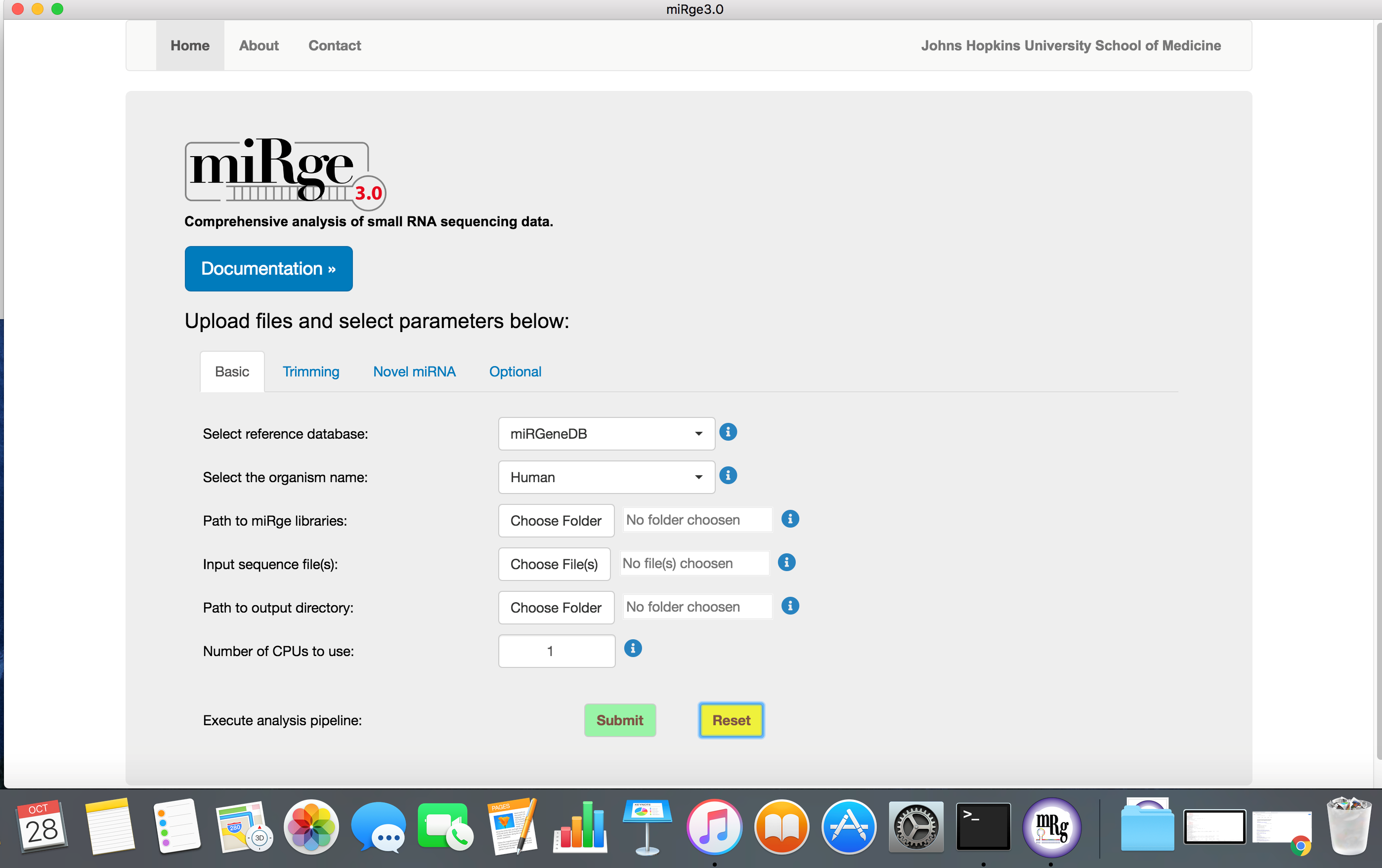
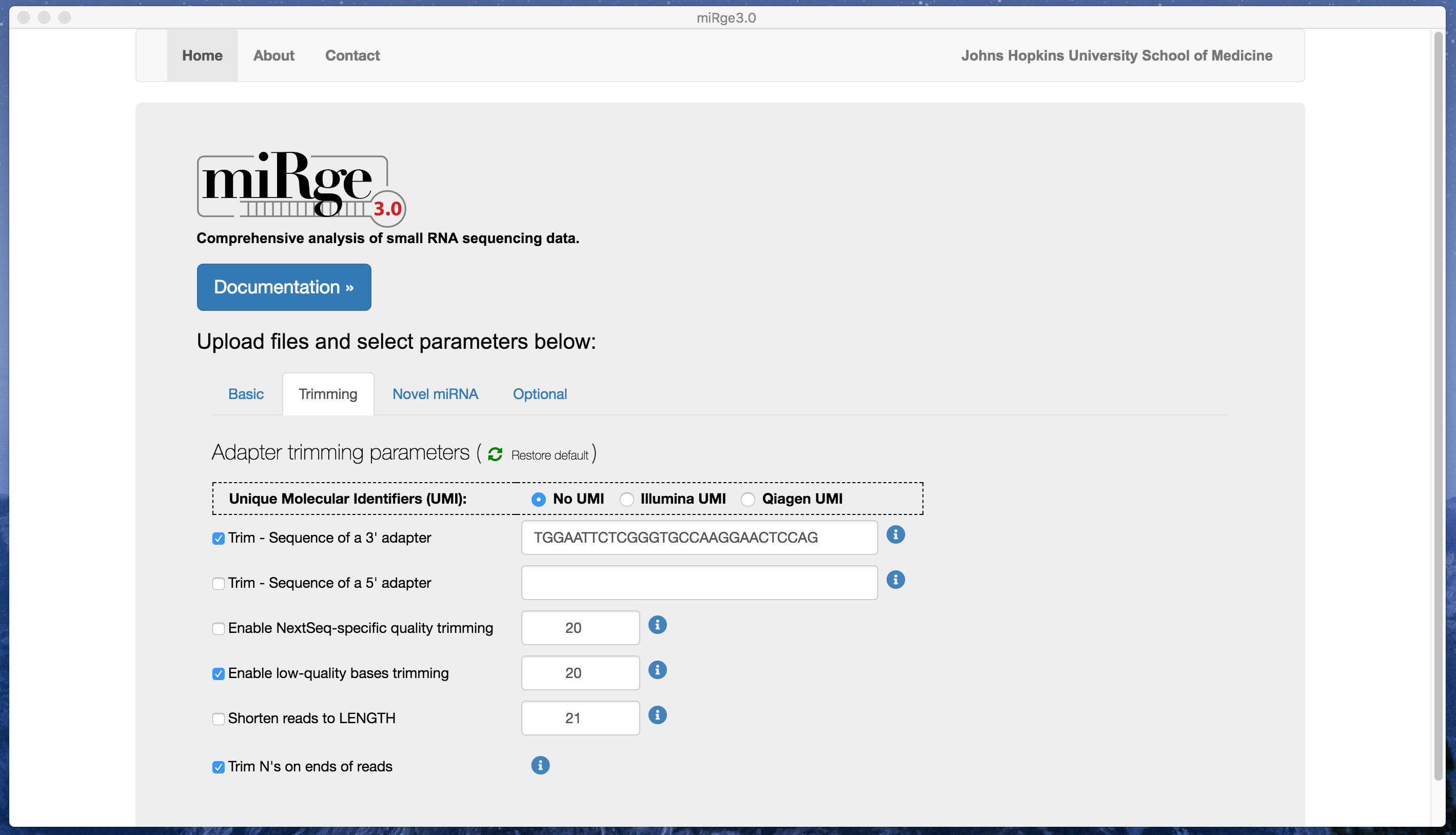
The software is easy to use with default parameters. The parameters are tabulated into four groups such as basic, trimming parameters, novel miRNA prediction and other optional parameters.
Screenshot with basic parameters
Screenshot with trimming parameters
Screenshot with novel miRNA predictions
Screenshot with other optional parameters